Ti:sapphire flexibility bridges a wide range of applications

Ultrafast-laser amplifiers have evolved into reliable, easy-to-use tools with ready access to a wide range of output characteristics. For many research applications, wavelength tunability is a necessary requirement that can now be conveniently achieved with fully computer-controlled amplifier systems, from seed-laser optical parametric amplifiers. Additional flexibility in pulse-width and energy can be obtained through open-system architectures. Because of their advantages, diode pumping and computer control are finding use in a growing range of spectroscopy applications.
Novel techniques
A common challenge in time-resolved pump-probe spectroscopy is to monitor the state-to-state distribution of a sparsely populated state created by the pump pulse. This population can be sparse simply because of the low sample concentration or because the state of interest is reached by a low probability event. Here, an important metric of the data quality is the signal-to-noise ratio.
Unfortunately, there are many types of pump-probe experiments in which simple, high signal-to-noise ratio (S/N) techniques such as LIF (laser-induced fluorescence) and MPI (multiphoton ionization) are not applicable. However, the simplicity and multiwavelength capabilities of the latest ultrafast-laser systems have enabled development of relatively complex techniques that can also deliver high S/N. A group led by Wolfgang Kiefer at the University of Würzburg, Germany (Institut für Physikalische Chemie) has been at the forefront in developing several of these techniques, including femtosecond time-resolved coherent anti-Stokes Raman spectroscopy (CARS), and population grating spectroscopy. Continuous-wave pumped amplified systems are an important tool for these investigations
In population grating spectroscopy, the pump pulse is split into two and recombined in the sample. Interference between these coherent wavefronts results in a spatially modulated pattern of laser illumination. When the pump wavelength is tuned to an absorption band in the sample, the result is a striped pattern of excited-state molecules; that is, a population gratingwhose lifetime is determined by the lifetime of the particular upper state involved.
The probe beam then interacts with this transient grating. Specifically, when the probe beam is near resonant with an absorption from the state(s) populated by the pump beam, the probability of elastic scattering increases significantly. But because the excited sample is arranged as a spatial grating, the elastically scattered light is in a beam whose direction is given by the phase-matching condition (see Fig. 1). A monochromator and CCD are set up to disperse and detect this diffracted beam. The lifetime of the transient state(s) can then be determined by measuring the signal strength as a function of the delay between the pump and probe beams. Moreover, the ability to change the probe-beam wavelength and disperse the scattered signal as a function of wavelength allows the population distribution of the transient state to be measured in some detail.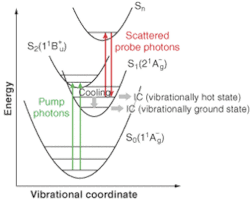
Kiefer and coworkers recently used this technique to study the dynamics of the S1 excited state of beta carotene, relevant for understanding the role of carotenoids in regulating energy transfer in the process of photosynthesis. The researchers used the pump pulse to excite a short-lived S2 excited state in beta carotene and then used the probe pulse to follow the evolution and decline of populations within the S1 state (see Fig. 2).
Electro-luminescent polymers
In a joint study, Art Epstein of the Physics and Chemistry Departments at Ohio State University (Columbus, OH) and Terry Gustafson’s group at the Center for Chemical and Biophysical Dynamics (CCBD) in the Chemistry Department at Ohio State University are studying fundamental properties of electro-luminescent polymers used to form organic LEDs (OLEDs), using a laser system funded by the National Science Foundation (NSF), with additional support from the university and the state of Ohio. In semiconductor OLED materials, an electron is promoted from the valence band into the conduction band and then moves along the polymer chain. A common polymer of this type is poly (p-phenylenevinylene) or PPV.
“Our research is aimed at revealing the relationship between structure and electron propagation,” Gustafson said. “Hopefully, this information will then prove useful to OLED designers, enabling them to produce polymers and devices that are brighter, more efficient, and more robust. In our work we use tunable ultrafast pulses to essentially optically dope the polymer; absorption of a photon excites an electron into the conduction band. As this propagates along the chain, we are interested in the accompanying changes (such as conformational changes) to the polymer chain.”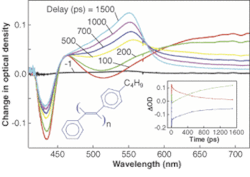
The team uses visible spectroscopy to study the evolution of the local electronically excited state, and longer-wavelength absorption to study the accompanying structural change via the vibrational spectrum. The studies include experiments with both polymer solutions and condensed-phase (thin-film) samples. This allows the researchers to study and isolate the intrachain properties from effects due to interchain interactions. By using chemically modified versions of PPV, the team is able to study how the local environment affects the propagation of the charge carrier. Gustafson noted that ultrashort pulses are necessary, as these processes occur on the subpicosecond timescale. He also noted that no single spectral measurement can reveal all the necessary information, so system flexibility is a critical advantage allowing his team to quickly switch between different types of spectral measurement (see “Optimizing design for spectroscopy applications,” above).
In a sample of phenyl di-substituted polyacetylene in toluene solution, absorption of the 400-nm pump pulse created transient species with unique absorption spectra (see Fig. 3). The sample was then interrogated with a broadband probe pulse at delays of 1 to1500 ps. The experiments were then repeated at different pump wavelengths. Combining these with other data revealed that intrachain structural changes allowed a short-lived initially excited state to evolve into a longer-lived excited state.
Photo-induced mutation
Members of Bern Kohler’s research group in the Department of Chemistry at Ohio State University use instrumentation in Ohio State’s CCBD to study polymers of a very different kind. The group uses femtosecond transient absorption and fluorescence upconversion spectroscopy to understand the evolution of excited electronic states created in DNA by UV light. These excitations, which are localized in the strongly UV-absorbing bases, can initiate photoreactions that corrupt the genomic information encoded by DNA. If left unrepaired, DNA damage can lead to cell death or cancer.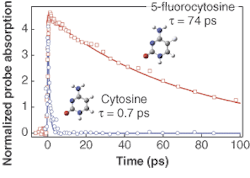
Nature has endowed DNA with remarkable photophysical properties to combat photodamage. Kohler and his coworkers have shown that single bases, the building blocks of DNA, have excited-state lifetimes of only hundreds of femtoseconds. In a process known as internal conversion, the DNA bases transform electronic energy into vibrational energy or heat on a subpicosecond time scale. This rapid nonradiative process allows electronic energy to be dissipated four to five orders of magnitude more rapidly than is possible by emitting fluorescent photons. The same mechanism is used by sunscreen molecules and confers significant photostability by eliminating electronic energy before it can initiate photochemical reactions.
Femtosecond pump-probe experiments on chemically modified bases provide insight into the nature of the nonradiative decay pathways (see Fig. 4). Cytosine, one of the four natural bases found in DNA, returns to its ground state in less than one picosecond (0.7 ps). A single atom substitution yields a 100-fold longer lifetime for the modified nucleobase, 5-fluorocytosine (74 ps). Experiments on modified bases like 5-fluorocytosine provide insight into the molecular mechanisms underlying photostability. Kohler concluded that the very short upper-state lifetimes of the natural DNA bases is a key factor behind the photostability of single DNA bases. Interestingly, some of this photostability is lost when bases are organized into polymers. Ongoing work at Ohio State by Kohler’s group investigates the new decay channels for electronic energy that are unique to assemblies of multiple DNA bases. ❏
Optimizing design for spectroscopy applications
An ultrafast laser system consists of several components-laser oscillator, amplifier, their pumps, and tunable OPA (optical parametric amplifier) modules. The exact combination depends on the intended applications. However, with these systems increasingly used as shared resources, a “typical” complete system offers multiple wavelengths and various levels of flexibility. The heart of this system is a modelocked Ti:sapphire laser oscillator pumped by a solid-state, continuous-wave (CW) 532-nm pump laser. This Ti:sapphire unit acts as the system’s master oscillator with a repetition rate of 70 to 90 MHz. The output of this oscillator is then fed into one or more amplifiers through a “pulse picker.” The amplifier is pumped by a pulsed Nd:YLF laser or by a 532-nm CW laser. Two distinct types of regenerative amplifiers are available. The first features high pulse repetition rate (up to 250 kHz) and low pulse energy (up to 5 µJ), like Coherent RegA 9000. The second type offers a much lower repetition rate (up to 5-10 kHz) but with very high pulse energy (up to 2.5 mJ or more), for example, Coherent Legend. In recent years Ti:sapphire amplifiers have been integrated with the oscillator and pump lasers into a single closed box for maximum operational simplicity, like Coherent Libra. Both types of systems can achieve pulsewidths shorter than 50 fs, a regime achievable only with Ti:sapphire.
About the Author
Marco Arrigoni
Marco Arrigoni is vice president of marketing at Light Conversion (Vilnius, Lithuania). He previously served as director of marketing at Coherent (Santa Clara, CA) from 2007 through 2023.