Quantitative Microscopy/Drug Discovery: Adding on deep-tissue, subcellular, quantitative phase imaging
While optical microscopy of live cells and tissues is a main source of insight for life science researchers, measuring the effects of drugs and diseases is highly demanding because it requires observation over hours or days. Indeed, drug research needs high-throughput, noninvasive, label-free techniques to make quantitative, nanoscale, and 3D tomographic functional measurements on live specimens. Ideally, optical imaging of cell-based assays should be sensitive enough to rapidly assess inhomogeneous characteristics cell-by-cell across large populations. And imaging of 3D specimens will ideally enable subduing of multiple scattering backgrounds and exhibition of strong sectioning to suppress out-of-focus light. But drug-screening methodologies have remained essentially unchanged since the introduction of the in vitro human cell line screen in 1990. And while existing methods provide information on the overall effects on cell viability, they are restricted to bulk measurements of large sample sizes, and thus cannot measure proliferation kinetics at the individual cell level.
Quantitative phase imaging (QPI) is a microscopy approach that delivers in all these areas. Because of their label-free, low-illumination power capabilities, QPI methods are nondestructive and thus suitable for stable, long-term investigation. In addition, they image large fields of view while simultaneously providing submicron resolution in 3D.
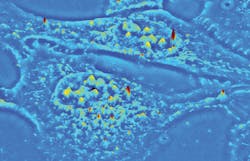
The established QPI technique known as spatial light interference microscopy (SLIM) excels at investigating thin, transparent samples such as single cells (see image above). Now, a new QPI technique called gradient light interference microscopy (GLIM) extends label-free and quantitative 3D tomographic imaging to optically thick, strongly scattering tissues with high resolution and remarkable contrast. Both have important implications for life sciences research and drug discovery.
Hastening drug discovery
Many drug discovery pipelines fail in early clinical trials,1 suggesting that more predictive biomarkers and preclinical cytotoxicity models are needed. Gaining a true understanding of cell proliferation to develop personalized adjuvant therapies calls for2-4 quantitative information about the effect of drugs on cell-based growth assays, as well as morphological and phenotypic changes at the single-cell level.
Preclinical research5-7 suggests that 3D tissue models are helpful for reproducing the complexity and heterogeneity of clinical tumors,8 including cell-cell interaction, hypoxia, drug penetration, response and resistance, and production/deposition of extracellular matrix. Tissue-like structures called organoids9-11 are used to recapitulate the phenotypes, function, and regulation of signaling pathways for in vivo tumor cells.12 Such optically thick, in vitro 3D structures (tissues, organoids, embryos, and model multicellular organisms) require live, long-term visualization and quantification on sub- and multi-cellular scales with high throughput.
Current methods
Live cells are notoriously difficult to observe under a microscope because of their high water content and lack of natural pigmentation. Current methods compensate by inserting contrast agents (fluorophores or dyes, including nanoparticles) directly or via genetic modifications.13-15 Optical microscopes with specialized light sources then excite the fluorophores and highlight the living structures and processes to enable measurement of their quantitative parameters. Imaging must be limited to short time intervals because the intense light energy causes photobleaching15, 16 and phototoxicity.17-21
Classical techniques such as phase contrast (PC)22 and differential interference contrast (DIC)23 microscopies can image live cells without exogenous agents because they create contrast from the subtle differences that result when light passes through the specimens. However, PC and DIC provide only qualitative measurements because intensity differences are not uniquely related to optical density differences in specimens.
Imaging thick 3D biostructures with any optical method involves loss of contrast because of multiple scattering.24 As thickness increases, multiple scattering generates background incoherence and exponentially decreases signal-to-noise ratio (SNR). The current methods25 for imaging live 3D biostructures are based on detection of spatial distribution of signal from fluorescence tags attached to features of interest.
Confocal microscopy (CM)26 uses a pinhole to reject out-of-focus background fluorescence, and allows optical sectioning in thicker samples than does conventional widefield fluorescence microscopy. But as imaging depth increases, the illuminating beam defocuses and passes less light through the pinhole, thus limiting CM’s penetration to about 100 µm in thick samples.27
Laser-scanning confocal microscopy (LSCM) provides better axial optical sectioning than spinning-disk confocal, as it performs point illumination of the sample and spatial filtering of the returning beam with a pinhole, which blocks extraneous light. Because LSCM uses very small apertures that reject a great deal of light, it requires high-intensity sources, bright fluorescence tags, and fixed specimens.
Two- and multiphoton microscopies28,29 reduce photobleaching and image deeply (~200–300 µm), while light-sheet and lattice light-sheet30,31 microscopies decouple the illumination and detection optical pathways to enable high-resolution axial imaging with low phototoxicity.32 Each of these techniques requires a specialized system and specific sample mounting, however, which prevents imaging of glass slides, petri dishes, and other common holders that allow specimens to maintain their natural structure.
Quantitative phase imaging
QPI33 overcomes the limitations of fluorescence microscopy. This rapidly expanding collection of alternative methodologies includes diffraction phase34 and digital holographic microscopies,36 quadriwave lateral shearing interferometry,37 ptychography,38 transport-of intensity equation-based techniques,39 optical diffraction tomography,40 and orientation-independent DIC.41 These methods provide contrast by quantifying changes in the wavefront (phase shift) when light propagates through the sample, and record it as pixel values in the generated image. Pixel intensity is proportional to the optical path length (OPL) change (that is, the physical thickness and the refractive index of the specimen), meaning QPI enables direct measurement of a specimen’s morphology42 and dry mass.43,44 Using QPI to measure phase shift across multiple angles of illumination or axial specimen positions,45-48 and solving the inverse scattering problems,49,50 can reveal the 3D structure of inhomogeneous specimens.
SLIM35 and GLIM60 are two QPI techniques that combine phase imaging (PC and DIC) with low-coherence interferometry and holography in a common-path geometry. They provide nanometer-level spatial phase sensitivity and high temporal stability as well. SLIM and GLIM were developed as add-ons for commercial PC and DIC microscope frames (respectively) in a 4f-optical relay design that provides high-fidelity optical field reconstruction between object and image planes with near diffraction-limited operation. Both make use of standard white-light illumination, which avoids the speckling that plagues the laser illumination-based QPI technique and improves optical sectioning, thanks to low coherence length of the light source.
Spatial and gradient light interference
Real-time, nondestructive SLIM (see Fig. 1) uses optical interferometry to reveal structure and dynamics with nanometer sensitivity in live cell cultures in 4D (3D time-series).51,52,35 In PC illumination, light beams emanating from the condenser ring pass through the sample and are collected in the back focal (Fourier) plane of the microscope objective.53 This scattered beam light is geometrically separated from the reference beam in the Fourier plane, and combined through interference in the image plane.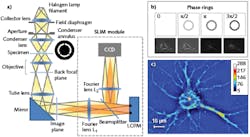
A phase plate in the Fourier plane advances the phase of the reference beam by 0.5π radians and attenuates its amplitude. Destructive interference generates a dark image for the dense portions of the cell with respect to the gray background. The SLIM module includes a one-to-one 4f relay system with a spatial light modulator (SLM) in the Fourier plane, and relays the microscope image plane with minimal aberrations (diffraction-limited) at a 1:1 ratio to a camera placed at its exit port. A phase mask ring, projected on the SLM screen at the same size, is conjugated with the back focal plane of the microscope objective. The SLM thus modulates the reference beam like a phase plate with variable thickness.
To create a quantitative phase image, the SLM shifts the phase of the reference beam by a fixed amount (0, 0.5π, π, 1.5π) and the camera captures the intensity image. A quantitative phase image is uniquely determined by combining the four frames and solving the field interference equations at each pixel. SLIM has successfully measured cell cycle-dependent growth and kinetics,54,55 enabled study of red blood cell structure,56 characterized the 3D structure of unlabeled live cells,57,48 and facilitated development of label-free cancer markers.58,59
GLIM (see Fig. 2), which complements SLIM, is designed to reject multiple scattering and enable strong optical sectioning for label-free, quantitative imaging of optically thick specimens.60 Its DIC illumination scheme includes two identical power cross-polarized beams shifted transversely by a condenser (Wollaston) prism with a distance smaller than the diffraction spot.53 After passing through the sample and a second Wollaston prism, the resulting interfering beams are sent into the GLIM module without passing through the analyzer.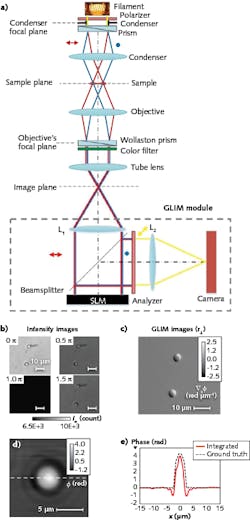
As with SLIM, the GLIM module incorporates a one-to-one 4f relay system with an SLM in the Fourier plane. The SLM active axis is aligned to the polarization direction of one beam and delays its phase by multiples of π/2 radians (quarter-wavelength OPL), while the other field remains unmodified. By accurately controlling the phase shift between the two beams, it can acquire four intensity images—which have the same incoherent background, but different coherent contributions—at the camera plane. Subtracting two pairs of images (sine and cosine) from each other retains the coherent component while withholding the multiple scattering.
The most important aspect of GLIM is that the two imaging beams are always equal in power, although they suffer equal degradation (that is, the same background noise) because of multiple scattering in the sample such that they interfere with high contrast, no matter how thick the specimen. Though it is new, GLIM has already been used by various research groups to visualize 3D time-lapse evolution of optically thick specimens, including zebrafish, C. elegans, and embryos.
Both SLIM and GLIM are available from Phi Optics in the form of modules that connect via C-mount to upgrade any commercial inverted optical microscope (10–100X magnification).
The modular units thus allow easy overlay and pixel co-registration with a microscope’s fluorescence channels to enable control and co-localization experiments based on fluorescence tags. Therefore, the SLIM and GLIM channels add to the existing fluorescence capabilities of a microscope seamlessly, offering unlimited combinations of 3D scanning and time series (see Fig. 3).
REFERENCES
For the complete list of references, please see www.laserfocusworld.com/home/article/14190428/may-2018-references.
About the Author

Dr. Gabriel Popescu, Ph.D.
Assistant Professor in the Department of Electrical and Computer Engineering
Professor Gabriel Popescu (1973-2022) received his B.S. and M.S. in Physics from University of Bucharest, in 1995 and 1996, respectively. He obtained his M.S. in Optics in 1999 and Ph.D. in Optics in 2002 from the School of Optics/ CREOL (now the College of Optics and Photonics), University of Central Florida. Dr. Popescu continued his training with the G. R. Harrison Spectroscopy Laboratory at MIT, working as a postdoctoral associate. He joined the University of Illinois at Urbana-Champaign (UIUC) in August 2007 as an Assistant Professor in the Department of Electrical and Computer Engineering and held a full faculty appointment with the Beckman Institute for Advanced Science and Technology. He was also an affiliate faculty in the Bioengineering Department. Dr. Popescu received the 2009 NSF CAREER Award, was the 2012 Innovation Discovery Finalist selected by the Office of Technology Management Office, UIUC, and was elected as the 2012-2013 Fellow of the Center for Advanced Studies at UIUC. Dr. Popescu was an Associate Editor of Optics Express and Biomedical Optics Express, and Editorial Board Member for Journal of Biomedical Optics.
Dr. Popescu passed away June 16, 2022.
Catalin Chiritescu
Chief Operating Officer, Phi Optics
Catalin Chiritescu is chief operating officer (COO) of Phi Optics (Champaign, IL).