Confocal filtering enables submicron detection with flow cytometry
Providing the ability to identify, sort, and count individual cells in heterogeneous mixtures, flow cytometers are valuable laboratory tools. Flow cytometry first transitioned from research labs to clinical application in the 1990s in response to the need for T-cell counting as a means of assessing HIV patients.1 Today, flow cytometers are commonly used for phenotypic characterization of blood cells, measurement of apoptosis markers, intracellular cytokine detection, and more.2 One of the newer and more exciting applications of flow cytometry is the measurement of submicron objects, especially extracellular vesicles. Because they are secreted from cells as a means of intercellular communication, extracellular vesicles are extremely interesting as biomarkers in clinical applications.3
The extremely small size of extracellular vesicles makes their detection challenging with traditional flow cytometers, though, because of the noise floor resultant from stray light in the collection path. Recently, the Wauben Research Group at Utrecht University (Utrecht, Netherlands), in collaboration with BD Biosciences Europe (Erembodegem, Belgium), developed a novel optical collection geometry to significantly increase the spatial resolution of a flow cytometer. The team first proposed the idea to optimize a confocal pinhole in the detection system as a means of reducing background noise and therefore increasing spatial resolution at the CYTO conference in 2017,4 and in 2020 published findings in the journal Cytometry Part A.5
Fundamentals of flow cytometers
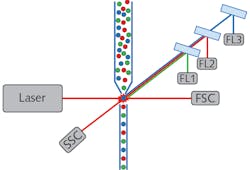
On the most simplistic level, a flow cytometer is a combination particle counter and fluorimeter, which enables sorting of cells and other small particles by physical and biochemical characteristics simultaneously. All flow cytometers consist of four base systems: fluidics, illumination, detection, and data analysis (see Fig. 1). Confocal filtering mainly affects the detection system, but it is helpful to understand the primary roles of the other systems as well. The fluidic system channels the individual cells or particles into a focused single file stream, allowing for each one to be investigated individually. The illumination source typically consists of one or more low-noise TEM00 lasers. The laser is focused into the particle stream to excite fluorescence and induce light scattering as each cell passes through the beam.
The detection system consists of two main parts: one to measure scatter intensity, and the other to measure fluorescence intensity at various wavelengths. The forward scatter component (FSC) is measured in line with the illumination path and the side scatter component (SSC) orthogonal to the illumination path. The FSC results from the laser beam diffracting around the scatterer, therefore correlating to the relative size of the cell or particle. The SSC, on the other hand, is dependent on the structure or granularity of the scatterer.
The fluorescence detection system of a flow cytometer is also configured to measure light orthogonal to the laser illumination path. Fluorescence is detected through a series of dichroic filters that direct the signal to wavelength-specific photodetectors, thereby allowing biochemical discrimination of cell types depending on whether they have affinity for particular fluorophores. It should be noted that in some flow cytometers, fluorescence is analyzed by a spectrometer instead of discrete filter-based detection channels.6
The data analysis system combines the physical data collected from the SSC and FSC detectors with the biochemical data collected by the fluorescence detection systems, allowing for a holistic counting and sorting of cells in a heterogeneous mixture. The information can be displayed as a one-dimensional histogram, two-dimensional scatter plot, or a three-dimensional contour plot, depending on the number of parameters the user wishes to compare. Additionally, many operators find it helpful to use information from one set of data points (e.g., SSC vs. FSC) to establish gating parameters on another set (e.g., different fluorescence detectors).
Increasing resolution by decreasing noise
Since the FSC is in line with the laser path, it is incredibly susceptible to background noise. Unfortunately, since the light scatters elastically, the FSC light is at the same wavelength as the illumination laser, rendering traditional optical filters useless. Over the years, instrumentation engineers have tried several different approaches to filtering out the laser light; the industry ultimately standardized on the use of an opaque obstruction placed in front of a collection optic as the most effective solution. This allows the laser light in the center of the optic to be blocked while collecting the off-axis FSC passing around the obstruction. This approach does an excellent job of filtering out direct laser light, but it does not do much to block off-axis stray light in the system, resulting in a background noise floor.
For most cell counting applications, the optical background noise is negligible, but as the particle size decreases, FSC intensity becomes too weak to be detected over the noise floor, making the analysis of submicron particles such as extracellular vesicles quite challenging. This is why the Wauben Research Group decided to tackle this problem by searching for an optimal pinhole in the optical path to act as a spatial filter. While the design is technically not truly confocal, due to the lack of conjugate pinholes, it produced a very similar effect since the individual scatterers themselves are conjugate to the pinhole. Figure 2 shows a schematic diagram of the optical layout of the FSC with the pinhole installed. From the diagram, it is easy to see how this quasi-confocal geometry can filter out stray light that would typically make its way around the obstruction and onto the photodetector.![FIGURE 2. The FSC quasi-confocal optical design shows the obstruction bar, focusing lens, and pinhole [6].](https://img.laserfocusworld.com/files/base/ebm/lfw/image/2020/09/2009LFW_chi_2.5f579332aafa5.png?auto=format,compress&fit=max&q=45?w=250&width=250)
In the article published in Cytometry Part A,5 Arkesteijn et al. explained how they tested this approach using a BD Influx Jet in Air sorter and a 20X Mitutoyo plan apo infinity-corrected objective with a 20 mm working distance to collect the FSC. They modified the system by placing an adjustable obscuration bar 7 mm in front of the objective, followed by a 50 mm focal length converging lens, which focused the FSC light through a pinhole placed 18 mm in front of the photodetector. Using 100-nm-diameter fluorescent microspheres, they tested 29 different obstruction and pinhole size combinations to find the combination that provided the best results: an 8 mm obstruction bar and a 200 µm pinhole. This was quantitated by looking at the FSC threshold, which they determined by examining the lowest number of events recorded during a 30-second background measurement.
The researchers went on to demonstrate the ability to detect not only microspheres, but also purified fluorescently tagged extracellular vesicles (~140 nm in diameter) using the same quasi-confocal optical design. They emphasized that even though this technique shows excellent results for measurement of purified extracellular vesicles, further testing is needed before use with heterogeneous biological samples can be validated. The authors also note that a “semi-quantitative analysis” was used during this research and they realize that a full Fourier optical analysis of the system in the future to understand the effects on collection efficiency.
Gaining momentum
Submicron applications of flow cytometry are rapidly gaining momentum, as evidenced by the fact that two of the plenary sessions at the 2020 Virtual CYTO conference were dedicated to aspects of it: “Subcellular Cytometry” and “Extracellular & Oceanic Cytometry.”
While the researchers’ results are still preliminary, confocal filtering shows great promise for enabling the analysis of extracellular vesicles and other submicron particles. As this technique and others like it continue to reduce optical background noise and increase the spatial resolution of flow cytometers, it is likely to open up new and revolutionary cytometry applications that have yet to be conceived.
REFERENCES
1. T. A. Fleisher and J. B. Oliveira, J. Clin. Immunol., 1239–1251 (2019).
2. A. Adan, G. Alizada, Y. Kiraz, Y. Baran, and A. Nalbant, Crit. Rev. Biotechnol., 37, 2, 163–176 (2017).
3. G. Van Niel, G. d’Angelo, and G. Raposo, Nat. Rev. Mol. Cell Biol., 19, 4, 213 (2018).
4. G. J. Arkesteijn, S. F. Libregts, E. N. Nolte-‘t Hoen, and M. H. Wauben, “Improved flow cytometric light scatter detection of submicron-sized particles by reduction of optical background signals,” 32nd Congress of the International Society for Advancement of Cytometry, Boston, MA (Jun. 10-14, 2017).
5. G. J. Arkesteijn, E. Lozano‐Andrés, S. F. Libregts, and M. H. Wauben, Cytometry Part A, 97, 6, 610–619 (2020).
6. B. Gefvert, Laser Focus World, 55, 2, 28–30 (2019); https://bit.ly/LFW-Cytometry.
About the Author

Robert V. Chimenti
Director, RVC Photonics LLC
Robert V. Chimenti is the Director of RVC Photonics LLC (Pitman, NJ), as well as a Visiting Assistant Professor in the Department of Physics and Astronomy at Rowan University (Glassboro, NJ). He has earned undergraduate degrees in physics, photonics, and business administration, as well as an M.S. in Electro-Optics from the University of Dayton. Over a nearly 20-year career in optics and photonics, he has primarily focused on the development of new laser and spectroscopy applications, with a heavy emphasis on vibrational spectroscopy. He is also very heavily involved in the Federation of Analytical Chemistry and Spectroscopy Societies (FACSS), where he has served for several years as the Workshops Chair for the annual SciX conference and will be taking over as General Chair for the 2021 SciX conference.