PLASTIC OPTICAL FIBERS: Stretching polymer opal fibers tunes structural color

CHRIS FINLAYSON, DAVID SNOSWELL, PETER SPAHN, and JEREMY BAUMBERG
Opal photonic crystals contain spheres of subwavelength diameter packed in regular arrays. Polymer opal fibers can now be produced that exhibit structural color based on the self-assembly of submicron core-shell particles. In both experiment and theory, these fibers are characterized by an internal substructure in which a concentric zone near the exposed surface develops particularly strong structural color. The fibers have a spectrum that is stretch-tunable across the visible region and are thus potential candidates for a novel range of nanomaterials and clothing fabrics that use strong structural color effects as a replacement for toxic and photodegradable dyes. These high-quality polymer opal fibers can now be produced in an industrially scalable extrusion process.1
Solvent-free manufacture
A low-cost technique to fabricate flexible opals has been developed using melting and shear-ordering of core-shell polymer nanoparticles.2–4 This produces low-defect flexible polymer face-centered-cubic (fcc) opal films with fundamental optical resonances tunable across the visible and near-IR regions by varying the precursor nanosphere size from 200 to 350 nm, and hence the resulting fcc lattice parameter. Such polymeric opals have intrinsic advantages over other self-assembling colloidal systems in that the structures formed are solvent-free and permanently formed as a mechanically robust solid.
In the lower refractive-index contrast regime associated with these polymer composites, color generation arises through spectrally resonant scattering inside a 3D fcc-lattice photonic crystal, as opposed to normal reflective iridescence based on Bragg diffraction.5 This principle is of fundamental interest in understanding the origins of structural colors and iridescence in natural opaline-type materials, such as those in minerals or in biological structures. In addition, one of the most attractive features of elastomeric polymer opals is the tunability of their perceived color by the bending or stretch modification of the (111) plane spacing.6 Such properties raise the possibility of producing finely drawn or extruded opaline fibers, using strong structural color effects as a replacement for toxic and photodegradable dyes in a variety of materials, such as clothing fabrics. In polymer opal films, ordering proceeds from the outer flat surfaces, which seed accumulation of (111) planes.7 However, recent work has now shown that it is also possible to produce opaline fibers by extrusion in a wire geometry.
The new fabrication technique uses shear-flow self-assembly of polymer particles into a 3D fcc lattice through extrusion at higher temperatures. The core-shell particle precursors are typically 200 to 300 nm in diameter and consist of a hard polystyrene (PS) core coated with a thin soft polyethylacrylate (PEA) outer shell (see Fig. 1). These particles are prepared using a multistage emulsion polymerization process. The material may be processed using a mini-extruder, which consists of two counterrotating metallic screws with adjustable speed in the range of 1 to 150 rotations per minute and adjustable temperature between 25°C and 250°C. Bulk quantities of the opal precursor materials are manually driven into the extruder, where they form a melt and are homogenized under the extreme shear forces provided by recycling through the screws. The overpressure generated then drives the shear-ordered granular material through a narrow-bore stainless steel die, producing thin opaline fibers.
The introduction of a small fraction of carbon nanoparticle dopants (about 0.1% wt) into the interstices of the photonic-crystal lattice does not disrupt the lattice quality but has been shown to result in a remarkable increase in the color saturation of the opals via spectrally-resonant scattering. Fibers may also be crosslinked by the addition of 2% wt of benzophenone and then curing in UV-A light. The final fiber products have sufficient mechanical robustness to allow them to be knitted into stretchable fabrics.
Stretch-tuning
The polymer opal fibers show significant changes in color as they are stretched. Color changes were visually and spectroscopically monitored in a 1000-µm-diameter red opal sample, showing a change from red through yellow/green, blue, and finally to a grayish color, as the strain e (the change in length divided by the original unstretched length) increased from 0 to 50% (see Fig. 2). These color changes are due to the decrease of the interplanar distance during stretching, as explained by Bragg diffraction. During the stretching of fibers, spheres within each plane parallel to the surface move apart but planes normal to the surface move closer to each other in order to keep the total volume constant, which causes the Bragg wavelength to shift to lower values. There is hence a roughly linear relationship of peak wavelength to strain, as expected from straining polymer multilayers.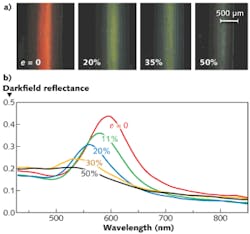
The darkfield reflectance for different strains reveals a sharp resonance (full width at half maximum of about 40 nm) in scattered light for the unstretched fiber, corresponding to the marked red appearance seen visually at near-normal incidence. As the fiber becomes increasingly stretched, the spectra are blue-shifted but also become notably broader in bandwidth as the ensemble of core-shell particles is transformed from fcc to a monoclinic lattice with strain. For strains above 50%, the spectra become very flat and broad, as expected from the pale gray appearance of the fiber.
The effect of stretching on ordering and coloring of fibers is reversible, given that the crosslinked fibers have not exceeded the elastic limit for e less than 50%. Similar color changes from red to blue/gray are also observed when fibers of other diameters are stretched. Typically the degree of spectral shift is -3 nm per percent of strain. Such fibers could be used in applications as diverse as textiles and photonic devices, for instance as matching filters surrounding cylindrical organic solar cells.
Substructure
One particularly interesting characteristic of these extruded polymer opal fibers is the substructure contained within the radial profile. From an image of the transverse cross section of a 2000 µm fiber, it is apparent that the interior of the fiber is not homogeneous (see Fig. 3). The substructure of the fibers generally consists of a series of concentric circular zones of different color. By studying the strength of the opaline resonance when collecting scattered light from different points on the fiber, it is found that the best ordering is found in the reddish zone just below the surface of the fiber. The outermost greenish zone, of depth 40 μm, is rather less well ordered (with a larger linewidth). This additional poorly ordered outer layer is not found for processed flat film geometries, and probably arises from the effect known as “die swell” as the fiber exits the cylindrical extrusion die.
To gain understanding of the mechanism by which sphere order develops through flow, a simple generic model for spheres is introduced.8,9 Through nonequilibrium interparticle dynamics, the interactions in a cylindrical geometry are chosen to model flow through a smooth circular pre-extrusion channel. The flow of polymer opals can be modelled as a granular system of particles, as there is no solvent to be considered. Each “particle” is represented as a soft sphere interacting through a radial pairwise potential with other spheres. The force between two spheres is characterized by a strong repulsion from the hard sphere core, combined with an attraction for spheres that are in contact to reproduce the “sticky” nature of the interaction.
In the simulation, ordered regions are observed developing at the surface boundary of the fiber. These initially grow locally along the face of the boundary, but eventually spread inwards once the order has developed to a large enough area to seed further templating inward. After sufficient time, the order is developed and a balance between ordered skin and disordered core is obtained.
REFERENCES
1. C.E. Finlayson et al., Opt. Exp., 19, 3144–3154 (2011).
2. T. Ruhl et al., Polymer, 44, 7625–7634 (2003).
3. O.L.J. Pursiainen et al., Appl. Phys. Lett., 87, 101902 (2005).
4. C.E. Finlayson et al., Adv. Mater., 23, 1540–1544 (2011).
5. O.L.J. Pursiainen et al., Opt. Exp., 15, 15, 9553–9561 (2007).
6. A. Kontogeorgos et al., Phys. Rev. Lett., 105, 233909 (2010).
7. O.L.J. Pursiainen et al., Adv. Mater., 20, 1484–1487 (2008).
8. L. Verlet, Phys. Rev., 159, 98 (1967).
9. L. Verlet, Phys. Rev., 165, 201 (1967).
Chris Finlayson has just moved to Cambridge to become a lecturer in physics at the Cavendish Laboratory; e-mail: [email protected]. David Snoswell is a research fellow in the NanoPhotonics Centre; e-mail: [email protected]. Peter Spahn is a group leader at the Deutsches Kunststoff-Institut, Darmstadt, Germany; e-mail: [email protected]. Jeremy Baumberg is director of the NanoPhotonics Centre at the Cavendish Laboratory, Cambridge, England, and also director of the Cambridge Nano Doctoral Training Centre; e-mail: [email protected].