Computational microscope achieves sub-atomic imaging resolution
Scientists at the University of Illinois (Champaign, IL) have built a computational microscope that can simulate the atomic and subatomic forces that drive molecular interactions. This imaging tool will streamline efforts to understand the chemistry of life, model large molecular systems and develop new pharmaceutical and industrial agents. The research is detailed in the journal Nature Methods.
RELATED ARTICLE: Computational microscopy approach can 'paint' tissue samples with light
The scientists combined two computational approaches used to simulate molecular interactions. The first, a nanoscale molecular-dynamics program known as NAMD, uses classical-mechanics methods to model the structure and simulate the behavior of hundreds of millions of individual atoms. The second program zooms in on the subatomic realm, simulating the interactions of protons, neutrons and electrons. Modeling at this quantum-mechanical scale demands a lot of computational power, so the researchers implemented a method for partitioning large molecules into classical- and quantum-mechanics regions. This allows them to focus their computational resources on small regions involved in critical interactions, such as the making or breaking of chemical bonds.
"We set it up so that researchers can easily choose how they will partition their own systems," said University of Illinois chemistry professor Zaida (Zan) Luthey-Schulten. "My own students are trying it out, and most of them are able to do it without much difficulty."
U. of I. physics professor Klaus Schulten developed NAMD at Illinois in 1995, combining it with visualization software, VMD, that enables researchers to watch large-scale molecular interactions unfold. Schulten, who died in 2016, equated this approach to "building a computational microscope."
The computational microscope is ideal for modeling structural traits and motions of large complexes. For example, in 2013, Schulten and his colleagues used NAMD to model the HIV capsid, which is made up of more than 1300 identical proteins that assemble into a cagelike structure that protects the virus until it enters a host cell. That simulation accounted for the interactions of more than 64 million atoms and required the use of the Blue Waters supercomputer at the National Center for Supercomputing Applications at the U. of I. The new study also made use of Blue Waters, this time to improve the resolution of the computational microscope.
In the new study, the research team at Illinois teamed up with QM experts Frank Neese, of the Max Planck Institute for Coal Research in Mulheim an der Ruhr, Germany; and Gerd B. Rocha, of the Federal University of Paraiba, in Joao Pessoa, Brazil.
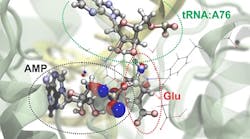
As a demonstration of the new approach, the researchers simulated the chemical behavior of transfer RNAs, molecules that play a key role in translating genetic information into proteins. Using NAMD, they modeled the overall molecular structure of tRNA at the moment that a special protein loads an amino acid to the tRNA. They partitioned two sites of the complex into regions requiring the more focused quantum mechanical approach.
The subatomic simulations of the interactions of the two regions allowed the team to run simulations of four different scenarios that would allow the tRNA to function as it does in the cell. Their simulations revealed that one of the four potential chemical pathways was more energetically favorable than the others and thus more likely to occur.
The molecular dynamics tools developed at the U. of I. are freely available to the public. This research was conducted in part at the National Institutes of Health Center for Macromolecular Modeling and Bioinformatics at the Beckman Institute for Advanced Science and Technology and the National Science Foundation Center for the Physics of Living Cells at the U. of I.
SOURCE: University of Illinois; https://news.illinois.edu/view/6367/630024
About the Author

Gail Overton
Senior Editor (2004-2020)
Gail has more than 30 years of engineering, marketing, product management, and editorial experience in the photonics and optical communications industry. Before joining the staff at Laser Focus World in 2004, she held many product management and product marketing roles in the fiber-optics industry, most notably at Hughes (El Segundo, CA), GTE Labs (Waltham, MA), Corning (Corning, NY), Photon Kinetics (Beaverton, OR), and Newport Corporation (Irvine, CA). During her marketing career, Gail published articles in WDM Solutions and Sensors magazine and traveled internationally to conduct product and sales training. Gail received her BS degree in physics, with an emphasis in optics, from San Diego State University in San Diego, CA in May 1986.